Authors
Sonia Rodriguez-Rodriguez,Tiana Huynh, Vi Lam, Andrew Chen, Carly Roleder, Tanya Siddiqi, and Alexey Danilov.
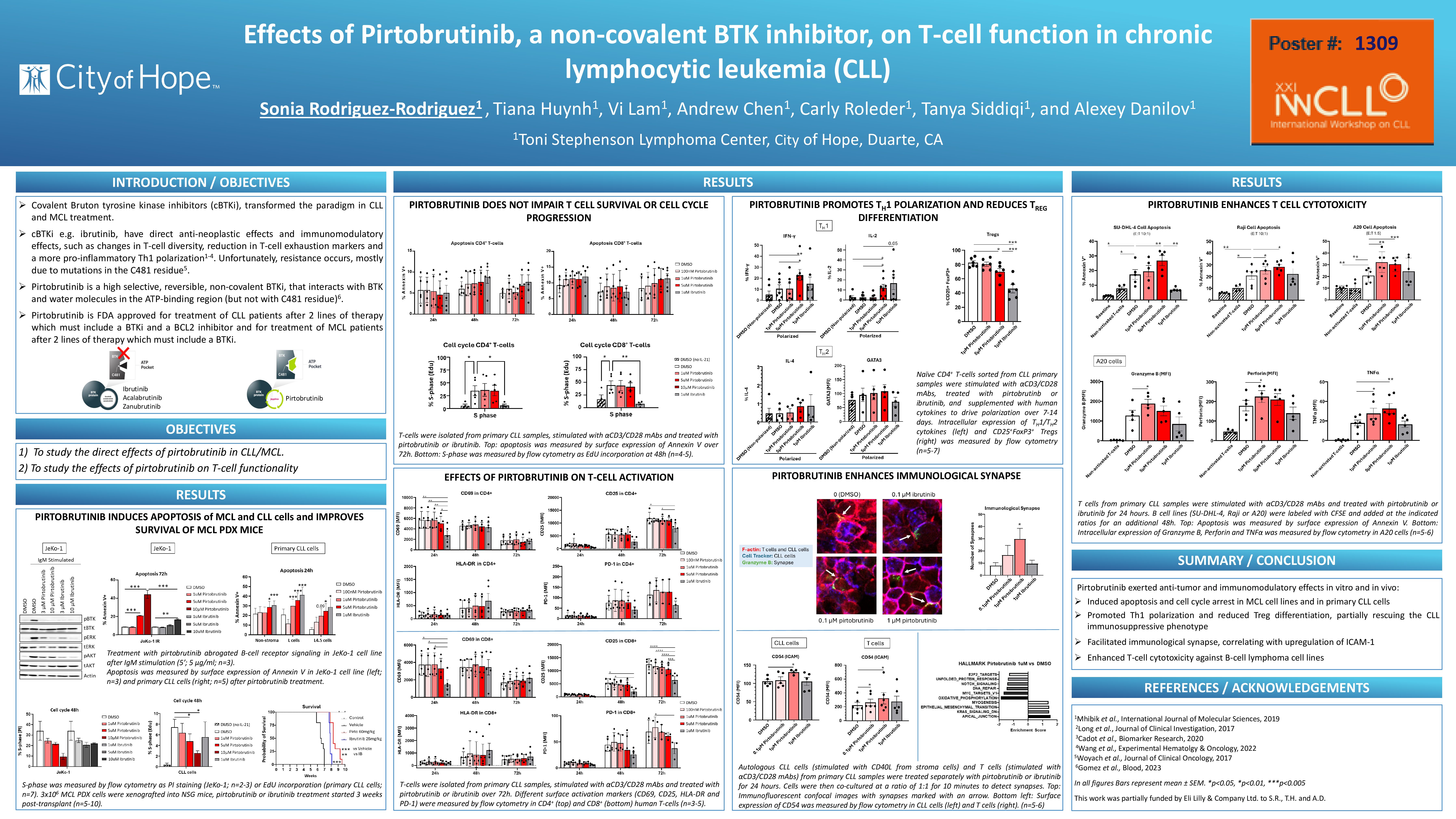
Covalent Bruton tyrosine kinase inhibitors (cBTKi), e.g. ibrutinib, transformed the treatment paradigm in CLL and Mantle Cell Lymphoma (MCL). cBTKi demonstrate direct anti-neoplastic effects. Additionally, cBTKi’s have clear immunomodulatory effects which are more prominent with ibrutinib compared with selective cBTKi’s acalabrutinib and zanubrutinib. However, resistance is inevitable, and in CLL is mainly driven by BTK mutations in the C481 residue. Patients who fail treatment with cBTKi have limited therapeutic options.
Pirtobrutinib is a highly selective, reversible, non-covalent BTKi, that has been FDA approved for treatment of patients with CLL and MCL who have previously received BTKi. However, the effects of pirtobrutinib on T-cell function are largely unknown. In our work we focused on studying the effects of pirtobrutinib in the T-cell compartment in CLL patients.
First, we demonstrated that pirtobrutinib abrogated B-cell receptor signaling in primary CLL cells and induced CLL cell apoptosis both off stroma and in CD40L expressing stromal conditions, which mimic the lymph node microenvironment. Pirtobrutinib treatment of mice inoculated with ibrutinib-resistant patient-derived MCL xenografts slowed expansion of MCL cells and extended survival by 1.5 weeks compare to ibrutinib and by 2.5 weeks compared with vehicle control.
Next, we measured the effects of pirtobrutinib on T-cell apoptosis, cell cycle and activation. CD3+ T-cells were isolated with DynabeadsTM FlowCompTM (ThermoFisher) from peripheral blood of CLL patients and stimulated up to 72 hours with αCD3/CD28 in the presence or absence of pirtobrutinib. Ibrutinib was used as control. Flow cytometry analysis showed that pirtobrutinib did not induce apoptosis or altered cell cycle progression in CD3+ T-cells. We observed downregulation of T-cell activation markers (CD69, CD25 and CD38) by ibrutinib, but not by pirtobrutinib which thus did not affect T-cell activation. Neither BTKi affected PD-1 expression in T cells. To further evaluate effects of pirtobrutinib with conducted RNA-Seq analysis of sorted naïve T-cells from healthy donors treated with the drug for 24 hours. Pirtobrutinib treatment led to ~80 significantly differentially expressed genes, compared with ~330 for ibrutinib. MYC and E2F2 targets, oxidative phosphorylation, unfolded protein response and DNA repair were the most significantly downregulated pathways following pirtobrutinib treatment.
Then, we used sorted naïve CLL-derived T-cells for in vitro polarization assays. Under Th1 polarization conditions, pirtobrutinib significantly upregulated secretion of IFN-γ and IL-2, to a greater extent than ibrutinib. Meanwhile, neither drug altered secretion of IL-4 and GATA3 under Th2 polarization conditions. Pirtobrutinib significantly reduced differentiation of Tregs in polarized conditions, albeit to a lesser degree compared with ibrutinib.
We next evaluated formation of immunological synapse. T-cells and CLL cells were treated with drugs for 24 hours, with T-cells simultaneously activated with αCD3/CD28, combined at a 1:1 ratio and analyzed by confocal microscopy. Interestingly, pirtobrutinib but not ibrutinib significantly enhanced immunological synapse formation. This increase correlated with upregulation of CD54 (ICAM-1) expression in both T-cell (T-cell activation and antigen uptake) and B-cell (cell-cell adhesion) populations, which may account for enhanced synapse formation. Next, we used SU-DHL4, Raji and murine A20 cell lines to conduct in vitro cytotoxicity experiments. Consistent with synapse data, pirtobrutinib but not ibrutinib enhanced T-cell mediated killing of neoplastic B-cells, accompanied by a significant increase of Granzyme B, Perforin and TNF-α secretion.
In conclusion, pirtobrutinib exerted anti-tumor effects in vitro and in vivo. Pirtobrutinib promoted Th1 polarization and reduced Treg differentiation, thus partially rescuing the CLL immunosuppressive phenotype, without compromising T-cell activation and expansion. Pirtobrutinib facilitated immunologic synapse formation and enhanced T-cell cytotoxicity. Our results justify continued investigation of pirtobrutinib for treatment of lymphoid malignancies.
Keywords : Pirtobrutinib, BTK inhibitor, T-cell
Please indicate how this research was funded. :
Please indicate the name of the funding organization.: