Authors
Martí Duran-Ferrer, Larry Mansouri, Guillem Clot, Ferran Nadeu, Sujata Bhoi, Lesley Ann Sutton, Panagiotis Baliakas, Sara Ek, Venera Kuci Emruli, Karla Plevova, Zadie Davis, Hanna Goransson-Kultima, Anders Isaksson, Karin E Smedby, Gianluca Gaidano, Anton W Langerak, Frederic Davi, Davide Rossi, David Oscier, Sarka Pospisilova, Maria Karypidou, Andreas Agathangelidis, Junyan Lu, Thorsten Zenz, Julio Delgado, Armando López-Guillermo, Paolo Ghia, Elías Campo, Kostas Stamatopoulos, Richard Rosenquist, José I. Martín-Subero.
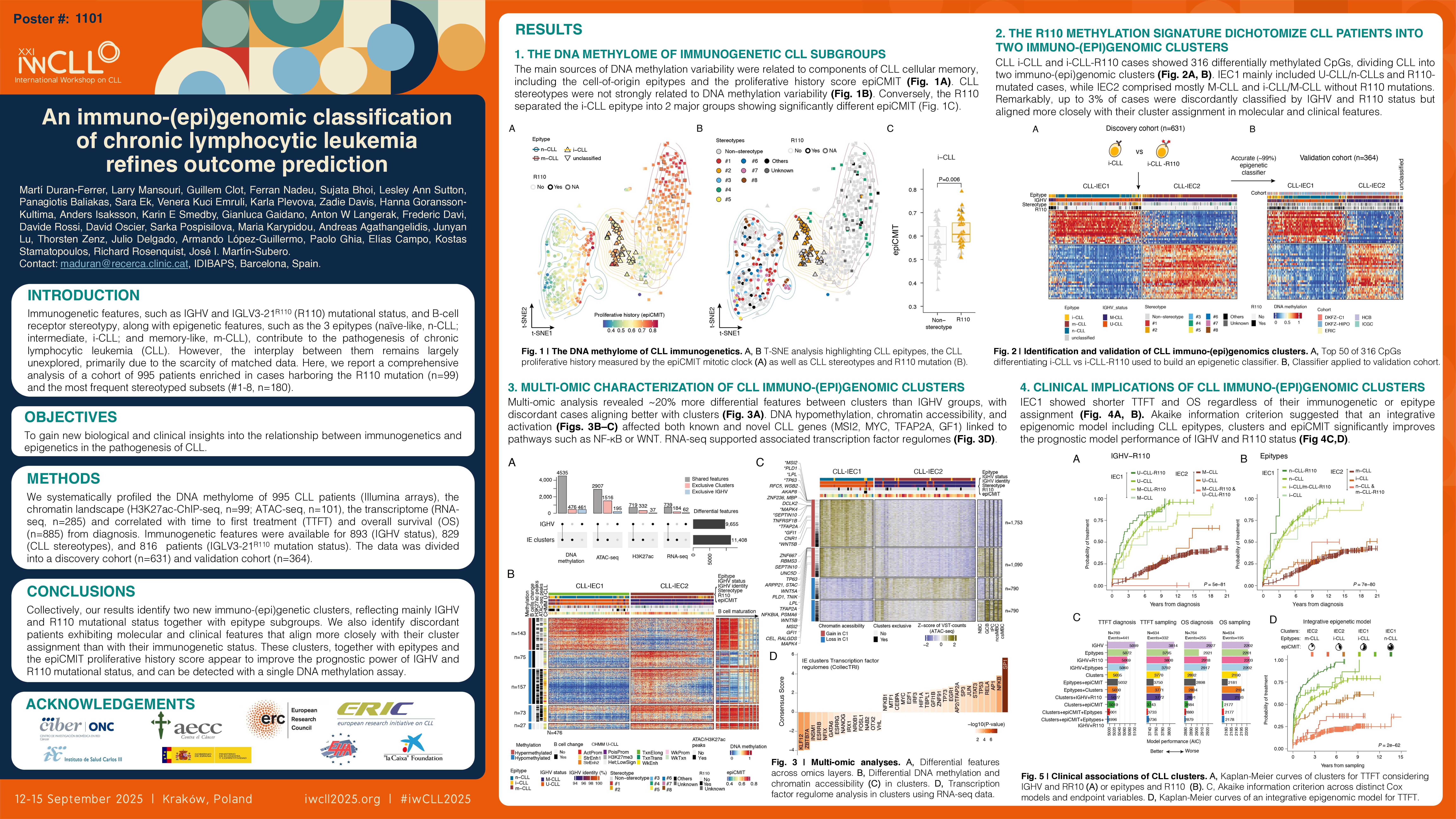
Immunogenetic features, such as IGHV and IGLV3-21R110 (R110) mutational status, and B-cell receptor (BcR) stereotypy, along with epigenetic features, such as the 3 epitypes (naïve-like, n-CLL; intermediate, i-CLL; and memory-like, m-CLL), contribute to the pathogenesis of chronic lymphocytic leukemia (CLL). However, the interplay between them remains largely unexplored, primarily due to the scarcity of matched data. Here, we report a comprehensive analysis of a cohort of 995 patients enriched in cases harboring the R110 mutation (n=99) and the most frequent stereotyped subsets (#1-8, n=180). We systematically profiled their DNA methylome (Illumina arrays, n=995), chromatin landscape (H3K27ac-ChIP-seq, n=99; ATAC-seq, n=101), transcriptome (RNA-seq, n=285) and correlated with time to first treatment (TTFT) and overall survival (OS) (n=885) from diagnosis.
Unsupervised analyses revealed that both subset and non-subset cases exhibited a similar DNA methylome, suggesting that stereotypy alone is not a major source of methylation variability. Nonetheless, poor-prognostic subset #2 and non-stereotyped cases carrying the R110 clustered together, indicating that the R110 mutation rather than subset #2 assignment was the molecular feature separating the i-CLL cases into 2 subgroups. Remarkably, i-CLL-R110 cases demonstrated a significantly higher (P=0.006) proliferative history measured by the epiCMIT score. Furthermore, i-CLL-R110 cases showed 316 differentially methylated CpGs, of which 154 were hypomethylated and located in regulatory regions (H3K27ac and ATAC-seq peaks).
Next, we used these 316 CpGs to develop (n=631/995) and validate (n=364/995) an epigenetic classifier that led to the classification of all CLL patients into two immuno-(epi)genomic clusters, named IEC1 and IEC2. IEC1 cases predominantly included IGHV-unmutated (U-CLL) and related U-CLL subsets as well as n-CLL and R110-mutated cases. Conversely, IEC2 cases included mainly IGHV-mutated (M-CLL), subset #4, m-CLL and i-CLL lacking the R110 mutation. However, we observed around 10% (84/884) of discordant cases compared to IGHV subgrouping, with 4% of all U-CLL (14/401) classified as IEC2, and 14% of all M-CLL (70/483) classified as IEC1. Considering both IGHV and R110 mutational status, the discordant cases were reduced to 3% (20/794, 5 M-CLL as IEC1, and 15 U-CLL as IEC2).
Subsequently, we performed a multi-omic analysis comparing both the newly defined clusters or IGHV subgroups. The clusters showed 5,011 differentially methylated CpGs, of which 476 displayed patterns independent of IGHV or epitype subgroups. Of these, 230 CpGs were hypomethylated and strongly enriched in regulatory regions, potentially contributing to the pathogenesis of IEC1 cases. In fact, we identified associations with TFAP2A, E2F, SP1 and MAZ transcription factor (TF) family members. From the chromatin perspective, we found 4,423 and 1,051 regions with differential chromatin accessibility (ATAC-seq) and activity (H3K27ac-ChIP-seq), respectively. Differential chromatin regions were predominantly related to higher accessibility/activation in IEC1 and targeted genes related to CLL pathogenesis such as MSI2 or TP63, and others less explored like GFI1 or TFAP2A. At the transcriptomic level, 923 genes were differentially expressed between clusters, with the majority overexpressed in IEC1 cases and overlapping with chromatin profiling results. IEC1 cases were enriched in gene signatures related to metabolism, cell cycle, NF-KB and WNT signaling, along with TF regulomes associated with these pathways, including SP1, NF-KB, TP53, TFAP2A, MYC or EGR1. Importantly, integrating all data types uncovered ~20% more differential features compared to IGHV subgroups, with discordant cases resembling cases of their cluster assignement and not their immunogenetic classification. Clinically, IEC1 cases were associated with a markedly shorter TTFT and OS, regardless of their immunogenetic and epitype subgroups, whereas C2 cases were independently associated with a longer TTFT (P=4e-89) and OS (P=4e-36). Akaike information criterion (AIC) demonstrated that clusters showed a better model performance than IGHV and R110 mutational status (for TTFT, 5,035 vs 5,069 and for OS, 2,892 vs 2,918, respectively), which became even more significant when including also epitypes and the epiCMIT (TTFT=5,001 and OS=2,880).
Collectively, our DNA methylation analyses identify two novel immuno-(epi)genetic clusters, reflecting mainly IGHV and R110 mutational status together with epitype subgroups. We also identify discordant patients exhibiting molecular and clinical features that align more closely with their cluster assignment than with their immunogenetic status. These clusters, together with epitypes and the epiCMIT proliferative history score appear to refine the prognostic power of IGHV and R110 mutational status, and can be detected with a single DNA methylation assay.
Keywords : immunogenomics, epigenetics, clinics
Please indicate how this research was funded. :
Please indicate the name of the funding organization.: