Authors
Dan Yang, Xiao Lu, Yeqin Sha, Luomengjia Dai, Ziyuan Zhou, Yi Miao, Yi Xia, Hui Jin,Jianyong Li,HuayuanZhu
Objectives
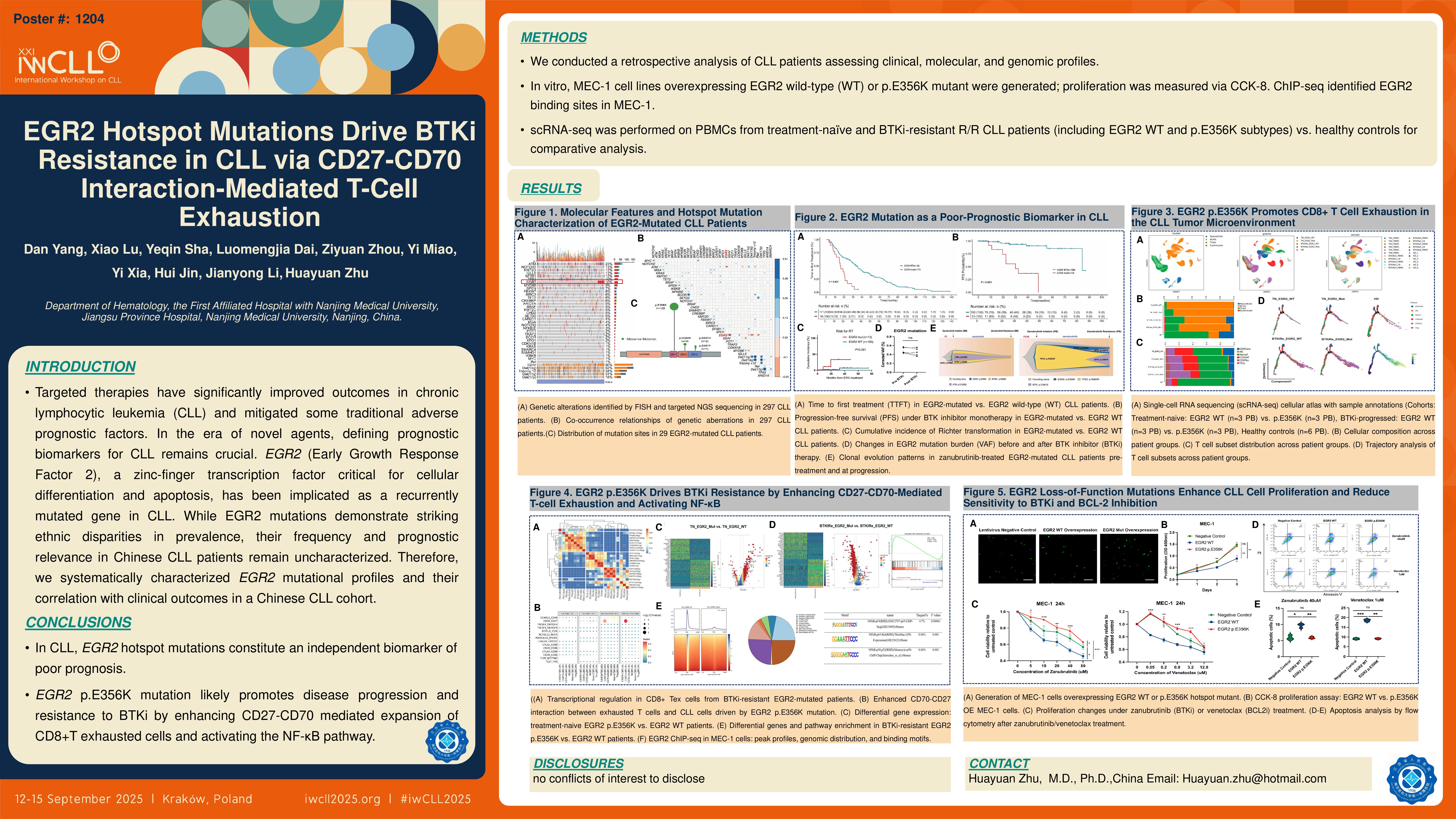
EGR2 (Early Growth Response Factor 2), a zinc-finger transcription factor critical for cellular differentiation and apoptosis, has been implicated as a recurrently mutated gene in chronic lymphocytic leukemia (CLL). However, the prevalence and prognostic relevance of EGR2 mutation in Chinese CLL patients remain unknown. Therefore, we systematically characterized EGR2 mutational profiles and their correlation with clinical outcomes in a Chinese CLL cohort. Furthermore, we explored the molecular mechanisms driven by EGR2 using single-cell RNA sequencing (scRNA-seq) technology to elucidate its role in CLL.
Methods
We retrospectively investigated CLL patients initially diagnosed and evaluated by next-generation sequencing (NGS) at Jiangsu Provincial People’s Hospital between January 2015 and January 2025. Comprehensive analyses were performed on clinical parameters, molecular characteristics, genomic mutation profiles, and therapeutic outcomes. Lentiviral vector infection was used to construct MEC-1 cell lines overexpressing EGR2 wild-type and EGR2 p.E356K hotspot mutant, and their proliferation was evaluated with CCK-8 assays. And we implemented scRNA-seq using peripheral blood mononuclear cells (PBMCs) from three treatment-naïve and three relapsed/refractory (R/R) patients resistanted to Bruton tyrosine kinase inhibitor (BTKi) including EGR2 wild-type and p.E356K mutant subtypes alongside six healthy controls for multi-dimensional comparative analysis. ChIP-seq to explore the binding sites of EGR2 in the CLL MEC-1 cell line.
Results
297 patients with newly diagnosed CLL meeting the inclusion criteria were analyzed. EGR2 mutations were detected in 9.8% of cases (29/297), with predominant variants located at p.E356K (75.6%, 22/29), p.H384 (13.8%, 4/29), and p.D411 (10.3%, 3/29). The median variant allele frequency (VAF) was 47%. Compared to wild-type counterparts, patients with EGR2 mutations exhibited: younger diagnosis age (50 vs 57 years, P< 0.001), advanced Binet staging (Stage C: 52.4% vs 25.2%, P=0.016), higher prevalence of monoclonal gammopathy (52.4% vs 18.0%, P< 0.001), increased co-occurrence of del(11q) (51.9% vs 15.4%, P< 0.001), ATM mutations (55.2% vs 19.0%, P< 0.001), and complex karyotype (CK) (31.0% vs 13.6%, P=0.029), significantly reduced del(13q) incidence (16.6% vs 45.0%, P=0.008). Furthermore, EGR2-mutated cases predominantly showed unmutated immunoglobulin variable heavy chain (IGHV) status (93.1% vs 44.1%, P< 0.001), the most frequent IGHV family was IGHV1 (37.9%, 11/29), enrichment in B-cell receptor (BCR) stereotyped subsets #1 (16.7%, 4/24) and #8 (16.7%, 4/24). Among 113 patients receiving continuous BTKi monotherapy, the EGR2-mutated cohort (n=13) demonstrated significantly shorter median progression-free survival (PFS) (30.1 vs 84.1 months; HR=5.69, P< 0.001) and elevated Richter transformation risk (23.1% vs 1.0%, P< 0.001) compared to wild-type patients (n=100). Furthermore, in five patients with EGR2 p.E356K hotspot mutations who developed resistance to BTKi monotherapy, longitudinal VAF monitoring showed no significant variations between baseline and post-resistance timepoints. Clonal evolutionary tracking revealed persistent dominance of the EGR2 p.E356K mutation as a major clone (VAF >45%) in two zanubrutinib treated patients, maintaining clonal prevalence both at initial diagnosis and upon acquired resistance. Furthermore, at the cellular level, the proliferation rate of EGR2 p.E356K-overexpressing MEC-1 cells was significantly higher than that of EGR2 wild-type-overexpressing MEC-1 cells (P < 0.01). scRNA-seq analysis further revealed that treatment-naïve patients with EGR2 p.E356K mutation had significantly higher peripheral blood T-cell proportions than EGR2 wild-type patients (13.24% vs 3.64%). Further T-cell subset analysis showed that EGR2 p.E356K-mutated patients had more exhausted T-cell infiltration than wild-type patients both pre-treatment (20.54% vs 10.53%) and after developing resistance to BTKi (31.02% vs 18.93%). CellPhoneDB analysis identified enhanced CD27-CD70 ligand-receptor interactions in mutated leukemic cells interacting with T lymphocytes, which further intensified upon BTKi resistance development. Differential gene expression analysis and Gene Set Enrichment Analysis (GSEA) identified NF-κB pathway hyperactivation in resistant patients. In the CLL MEC-1 cell line, ChIP-seq analysis showed 32.83% of binding peaks reside in promoter regions. EGR2 was found to interact with RelA (p65) binding motifs in these peaks, indicating its potential role in activating the NF-κB pathway via direct RelA binding.
Conclusions
In CLL, EGR2 hotspot mutations constitute an independent biomarker of poor prognosis and the EGR2 p.E356K mutation likely promotes disease progression and resistance to BTKi by enhancing CD27-CD70-mediated expansion of CD8+T exhausted cells and activating the NF-κB pathway.
Keywords : chronic lymphocytic leukemia, EGR2, Bruton tyrosine kinase inhibitor
Please indicate how this research was funded. : This work was supported by the National Natural Science Foundation of China (Grant No. 82170166) , the Suqian Science and Technology Program(Grant No.KY202305)and the Specialized Diseases Clinical Research Fund of Jiangsu Province Hospital (Grant No.DL202406).
Please indicate the name of the funding organization.: National Natural Science Foundation of China, Suqian Science and Technology Bureau, Jiangsu Province Hospital