Authors
Anja C. Rathgeber, Stacey M. Fernandes, Shuqiang Li, David M. Dorfman, Lars Bullinger, Mattew S. Davids, Jennifer R. Brown, Kenneth J. Livak, Leif S. Ludwig, Catherine J. Wu and Livius Penter.
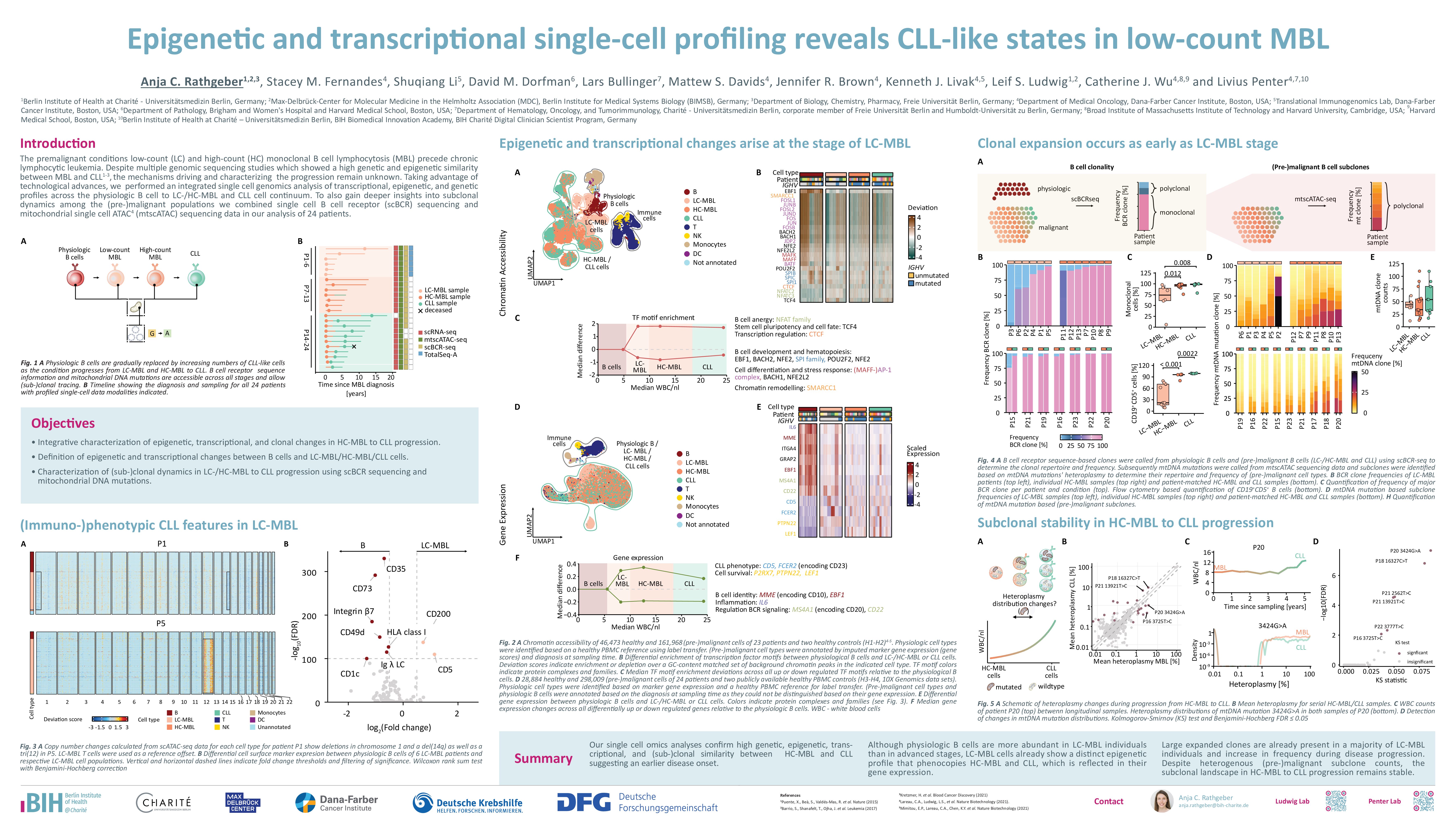
Chronic lymphocytic leukemia (CLL) is preceded by low-count (LC-) and high-count monoclonal B cell lymphocytosis (HC-MBL). As the precise order of events underlying initial clonal B cell expansion and subsequent leukemic transformation – including epigenetic and transcriptomic reprogramming – have not been fully characterized, progression from MBL to CLL remains difficult to anticipate clinically. Resolving CLL-initiating events would facilitate the prediction of progression from MBL to overt leukemia and inform the discovery of novel therapeutic strategies. Here, we performed a single-cell multi-omics characterization of MBL and CLL at the transcriptional, epigenetic, and proteomic levels. We performed lineage tracing with natural genetic barcode analysis of B cell receptor (BCR) sequences, mitochondrial DNA (mtDNA) mutations, and copy number variations (CNV), and further augmented this analysis through longitudinal tracking of MBL/CLL subclones to provide a comprehensive map of phenotypic and genetic evolution from LC- via HC-MBL to CLL.
The study cohort comprises 34 CD19+ enriched peripheral blood mononuclear cell (PBMC) samples from cases of LC-MBL (n=6), HC-MBL (n=7), and CLL (n=11), including paired HC-MBL and CLL specimens of 10 individuals (median follow-up time: 1,926 days, range: 916–2,859 days). We obtained 301,853 high-quality single-cell transcriptome (RNA-seq) and BCR profiles, along with 168,533 single-cell chromatin accessibility (ATAC-seq) and mtDNA mutational profiles. Based on a healthy PBMC reference and marker gene expression, we categorized cells into the major immune subsets of B cells, T cells, NK cells, and monocytes. We annotated the B cell compartment as physiologic or malignant based on BCR clonotype dominance (scRNA-seq) or distinct chromatin accessibility profiles of MBL/CLL (scATAC-seq).
While physiologic B cells exhibited conserved epigenetic profiles, MBL/CLL cells formed distinct, case-specific gene expression and chromatin accessibility clusters. Within this variability, MBL/CLL cells consistently exhibited altered activity of transcription factor binding motifs in comparison to physiologic B cells, such as EBF1, SMARCC1, AP-1, and BACH. These changes were already detectable at the stage of LC-MBL and persisted throughout the MBL/CLL transition. They were mirrored at the transcriptomic level, which showed evidence for the acquisition of a CLL-like phenotype starting at the LC-MBL stage. This CLL-like transcriptomic state reflected loss of B cell identity, inflammation, and dysregulation of cell survival due to altered expression of genes such as CD5, FCER2, MME, EBF1, IL6, and MS4A1. Together, epigenetic and transcriptional reprogramming is an early event in MBL initiation, and core changes remain stable throughout the progression to CLL.
Given the presence of CLL-like epigenetic and transcriptional states in LC-MBL, we explored its immunophenotypic and genetic features. BCR repertoire analysis demonstrated that LC-MBL retained only a small residual polyclonal B cell compartment (median: 25% polyclonal; range: 3-95%), which was consistent with the low percentage of CD19+ CD5- non-MBL cells on flow cytometry (median: 21%; range: 0.3-91%). Further, epigenetically defined LC-MBL cells exhibited a CLL-like surface immunophenotype, characterized by the overexpression of markers such as CD5 and CD200 or down-regulation of CD49d, Integrin β7, CD73, and CD35. Notably, in 2 of 6 LC-MBL cases, we found large-scale chromosomal alterations, such as trisomy 12. Together, molecular hallmarks of CLL emerge in LC-MBL at the genetic and surface marker level.
Finally, we investigated the relationship between epigenetic and transcriptional stability during the MBL/CLL transition and clonal evolution. While most MBL/CLL populations were characterized by a monoclonal BCR, we reasoned that mtDNA mutation analysis would further resolve (sub)clonal hierarchies with high resolution. Indeed, we found a median of 33 subclones per MBL/CLL (range: 4-90). The majority of HC-MBL subclones (median: 6 per case; 73% of all clones) were stable during progression to CLL. More granular analysis of heteroplasmy distributions – quantified via variant allele frequency of mtDNA mutations – further demonstrated subclonal persistence and stability. These results indicate that HC-MBL/CLL progression is predominantly driven by disease proliferation rather than the emergence or selection of novel clones.
In summary, we find that epigenetic and transcriptomic transformation toward a CLL-like phenotype occurs already in LC-MBL and remains stable throughout the HC-MBL/CLL transition. This supports a model in which core CLL-like states are acquired early and together with patient-specific cell states drive leukemogenesis. LC-MBL is a biologically heterogeneous and thus far under-characterized premalignant condition that warrants deeper investigation to uncover biomarkers and potential therapeutic targets for the prevention of progression to CLL.
Keywords : Leukemic transformation; Single-cell multi-omics; Clonal expansion
Please indicate how this research was funded: ASH-Scholar Award;
CLL P01 CA206978 grant
Please indicate the name of the funding organization: American Society of Hematology (ASH);
NIH/NCI