Highlighting a crucial role for EGR2 mutations in CLL pathogenesis and BTKi resistance (1MB pdf)
Authors
Alessia Morabito, Jessica Bordini, Chiara Lenzi, Michela Frenquelli, Lydia Scarfò, Alessandro Campanella, Paolo Ghia.
Background
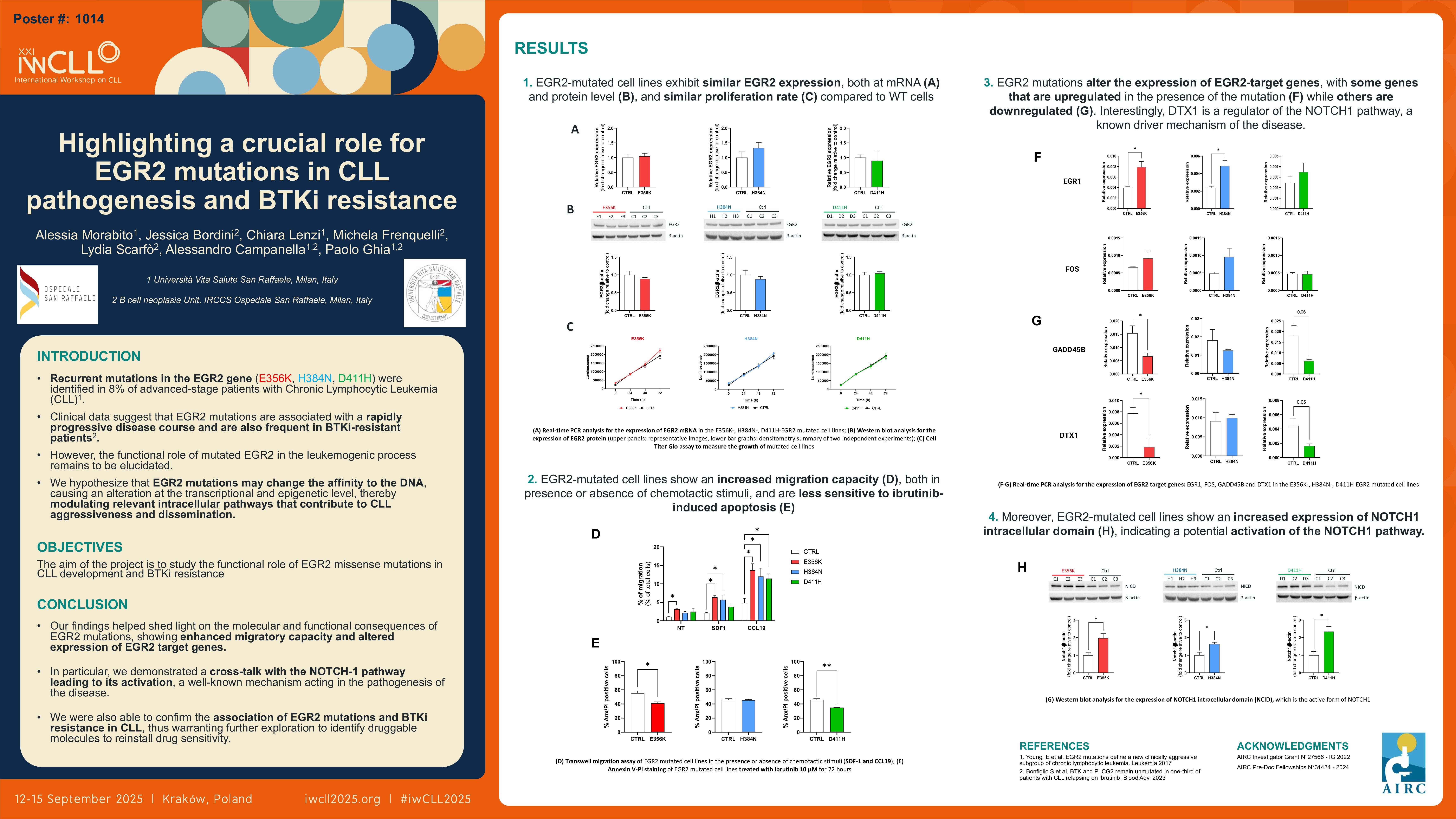
Chronic lymphocytic leukemia (CLL) is a B-cell malignancy characterized by a tissue involvement of both primary and secondary lymphoid organs, as a result of a complex intertwining between microenvironmental stimuli and genetic defects. We previously reported recurrent mutations in the EGR2 gene in 8% of advanced-stage patients with CLL, that associated with distinctive biological and clinical features of poor prognosis. EGR2 encodes for a transcription factor, activated in B cells through ERK phosphorylation upon BcR stimulation. Clinical data suggest that EGR2 mutations are associated with a rapidly progressive disease course characterized by shorter time to first treatment and overall survival. Interestingly, we also reported EGR2 mutations in patients becoming refractory to continuous BTK inhibitors, namely ibrutinib, particularly in association with BTK mutations. However, the precise role of mutated EGR2 in the leukemogenic process and in the occurrence of resistance to BTKi remains poorly explored, with recent evidence indicating an impact on the transcription of EGR2 target genes. Here, we are providing initial support to the hypothesis that EGR2 mutations may cause a gain of function of the transcription factor activity of the gene, thereby modulating relevant intracellular pathways that contribute to CLL aggressiveness, dissemination and drug resistance.
Aims
We aim at understanding the functional role of EGR2 mutations in CLL development and progression.
Methods
We engineered the MEC-1 CLL cell line using CRISPR/Cas9 technology to generate EGR2-mutated cells, selectively harboring each of the three recurrent missense mutations found in patients: E356K, H384N, and D411H. We analyzed the effect of EGR2 mutations by studying in vitro cell proliferation, transwell migration in the presence or absence of chemokines, expression of EGR2 target genes, and sensitivity to the BTK inhibitor Ibrutinib.
Results
We obtained three distinct EGR2-mutated clones for each missense mutation, along with control clones. All cell lines exhibited comparable expression of EGR2 both at mRNA and protein levels, as well as similar proliferation rates. Notably, EGR2-mutated cell lines demonstrated an increased migration capacity in the presence of SDF-1 and CCL19, chemotactic stimuli crucial for homing of malignant cells into bone marrow and lymph nodes. Gene expression analysis revealed that the mutated clones exhibited a differential expression of known EGR2 targets compared to controls, with certain genes (e.g. EGR1 and FOS) upregulated and others (e.g. GADD45B and DTX1) downregulated. Furthermore, EGR2-mutated cell lines had elevated levels of cleaved NOTCH1 protein, indicating a potential interaction between the EGR2 and NOTCH1 pathways, the latter known driver of the disease. Lastly, in line with our previous observation in refractory patients, the EGR2-mutated cell lines demonstrated reduced sensitivity to apoptosis induced by the BTK inhibitor Ibrutinib, in contrast to the control clones.
Conclusion
Our model, consisting of EGR2-mutated cells, shows an enhanced migratory capacity and altered expression of EGR2 target genes in the presence of each of the 3 missense gene mutations. In particular, we were able to demonstrate a cross-talk with the NOTCH-1 pathway leading to its activation, a well-known mechanism acting in the pathogenesis of the disease, being frequently altered in patients with CLL and associated with poor outcome. As originally reported by us in patients refractory to Ibrutinib, also in the mutated cell lines, we were able to confirm the association of EGR2 mutations and BTKi resistance in CLL; further exploiting this model may help shed light on potentially druggable molecules and/or pathways that will overcome or prevent resistance.
Keywords : leukemia, gene mutation, BcR signaling
Please indicate how this research was funded. :
Please indicate the name of the funding organization.: